








LCHってどんな病気?Ver.2 2022/10
ランゲルハンス細胞組織球症(Langerhans cell histiocytosis:LCH)
Frequently Asked Questions(FAQ)
はじめに
LCHは、さまざまな症状がでて、さまざまな経過をたどる、まれで不思議な病気です。さまざまなところに病変がでてくるため、患者さんは整形外科や耳鼻科・脳外科・皮膚科・呼吸器科・歯科などさまざまな科を受診します。医学部では主に小児科で習いますが、病気がめずらしく教科書の片すみにしか書かれていないため、この病気に詳しい医師はあまりいません。その結果、なかなか診断がつかず、診断がついても適切な治療がされないことがあります。LCHはまさに“orphandisease”(みなしご病)です。ここでは、最新のデータに基づきLCHについて解説します。
LCHという病名は?
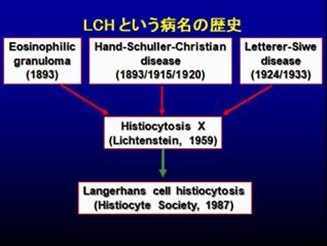
LCHには、以前、レテラー・ジーべ病(Letterer-Siwe)、ハンド・シューラー・クリスチャン病(Hand-Schuller-Christian)、好酸球性肉芽腫(eosinophilicgranuloma)の3つの病名がありました。LCHの症状や経過があまりにさまざまなので、別々の病気だと思われていたのです。これらの原因がいずれも組織球(ヒスチオサイト)によることがわかり、まとめてヒスチオサイトーシスX(histiocytosisX)と呼ばれるようになりました。そして、その組織球がランゲルハンス細胞であるということが判明し、1987年にランゲルハンス細胞組織球症(LCH)という病名が確立しました。
今では、病気の部位がひとつの臓器(単一臓器型)か、二つ以上の臓器(多臓器型)かで分けます。さらに、単一臓器型は、単独病変か多発病変かで、多臓器型は、リスク臓器(RO:後述)に病変があるかどうかで分けます。
| 現在の分類 | 病変の臓器 | 過去の呼び名 | |
|---|---|---|---|
| 単一臓器型 | 単独病変 | 多くが骨(皮膚やリンパ節もあり)成人では肺もあり | 好酸球性肉芽腫 |
| 多病変 | |||
| 多臓器型 | リスク臓器病変なし | 骨と皮膚など | ハンド・シューラー・クリスチャン病 |
| リスク臓器病変あり | 皮膚と肝臓、脾臓など | レテラー・ジーベ病 | |
どのくらい患者さんがいて、どんな人に多いの?
日本ではLCHにかかる子どもの数は年に数十人です。多臓器型は1歳未満に多く、ほとんどが3歳未満です。一方、単一臓器型は幅広い年齢ででてきます。患者さんの70~80%は子どもですが、成人の患者さんもあります。 成人では診断されていない患者もあると考えられます。子どもでは、男児にやや多くみられます。
原因はなんですか?
ランゲルハンス細胞は白血球の一種で、組織球と呼ばれる仲間に属します。元々は血を作る畑:骨髄から出てきたものです。皮膚や気管など外界と接するところにいて門番の役目をしています。体に異物が入ってくると、その異物を食べて、サイトカインと呼ばれる免疫を活性化するホルモンのようなものを出します。つまり、周りに「敵が来たぞ!戦闘態勢に入れ!」と警戒警報を鳴らします。そして、リンパ節に移動して、それがどんな異物かを免疫の司令官であるリンパ球に伝える働きをします。そのランゲルハンス細胞が、何らかのきっかけで、皮膚や骨・リンパ節などに異常に集まって病変を作るのがLCHです。LCHの病変部位には、ランゲルハンス細胞が鳴らす警戒警報に反応して、リンパ球や好酸球、マクロファージ、破骨細胞様多核巨細胞(骨などを融かす働きがある細胞)などが集まっています。これらの細胞は互いに刺激し合って、高度の炎症が生じます。つまり、病変部は「戦場」となり、組織は爆撃を受けたように破壊され、さまざまな症状が出ます。
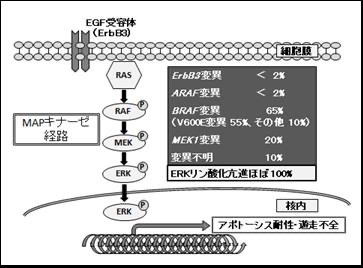
2010年に、異常に増えているランゲルハンス細胞(LCH細胞)にBRAF遺伝子の変異(BRAF V600E)が見つかりました。この変異は、悪性黒色腫や甲状腺がん、大腸がんでもよく見られるもので、発がん性の遺伝子変異といわれるものです。BRAF蛋白は、細胞質にある蛋白で、細胞の外からやってきた信号を核に伝えるMAPキナーゼ経路というところにあります。その後、次々に、MAPキナーゼ経路にある、MAP2K1(MEK1)遺伝子などに変異がある例が見つかりました。これらの遺伝子に変異が起こると、MAPキナーゼ経路の信号が異常に強く伝わる(ERKリン酸化亢進)ようになります。その結果、LCH細胞は生き続ける(アポトーシス耐性)ようになったり、リンパ節に移動しなくなったり(遊走不全)して、病変部に止まり集まります。LCHのほぼすべての患者さんで、MAPキナーゼ経路の遺伝子に異常が生じていることから、LCHは、白血球の一種である組織球が、「がん」細胞に似た状態であることが明らかになりました。しかし、LCH細胞では遺伝子変異はただ一つだけしか持っていません。また、自然に治ることがあります。これらのことは、通常の悪性度の高い「がん」とは大きく異なります。
これらの知見から、LCHは「炎症性骨髄腫瘍」という概念でとらえられています。
LCHに「がん」が合併することはありますか?
LCHの発症前・発症時・発症後に、急性白血病やリンパ腫などの悪性腫瘍を合併するとことがあります。LCH発症後に合併する悪性腫瘍の大部分は、放射線治療またはエトポシド(VP-16)という抗がん剤治療によるものと考えられます。
LCHは遺伝しますか?
LCH患者さんの100人に1人は家系内にLCHの患者さんがいるといわれています。また、一卵性双生児で一方がLCHの場合もうひとりもLCHになる率は高いといわれています。このことからすると、LCHになりやすいかどうかは、ある程度、遺伝的な要素があると考えられます。しかし、明らかな遺伝性の病気ではありません。
どんな症状がありますか?
子どもの単一臓器型の場合、ほとんどは骨病変で、皮膚やリンパ節に病変がみられる例も少数あります。多臓器型の場合、皮膚と骨病変の頻度が高く、肝、脾、肺、胸腺、骨髄などさまざまな臓器にも病変がみられます。
初発症状として、単一臓器型では骨が腫れる、骨の痛み、発熱が多く、多臓器型では皮膚のぶつぶつ、骨が腫れる、発熱、リンパ節が腫れる、肝臓や脾臓が腫れるといった症状が多くみられます。以下、病変の部位別に症状を示します。
-
骨病変
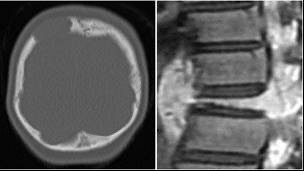
頭の骨に最も多くみられます。あばら骨や腰骨、背骨、あごの骨、手足の骨にもみられます。頭の骨の場合、こぶのように腫れてぷよぷよとし、その後、中心部がへこんでクレーターのような状態になります。頭をぶつけたところからでてくることがあります。足や骨盤の骨の場合、痛みで足を引きずることがあります。顎の骨の場合、歯が抜けることがあります。背骨の場合、骨の周りが腫れて神経を圧迫しヘルニアのような症状がでることがあります。目の周りの骨の場合、目がとび出たり視力がおちたりすることがあります。単純レントゲンでは骨が融けたようにみえます。打撲した部位に病変が生じることがあります。
皮膚病変
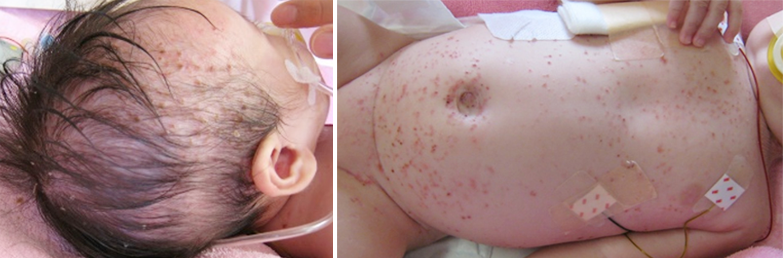
頭や脇、股などに脂漏性湿疹、体幹などにあせも様または出血斑様の小丘疹がみられます。アトピー性皮膚炎やおむつかぶれと間違われることもあります。
-
リンパ節病変
首のリンパ節が腫れることが多くみられます。
-
耳病変
なかなかよくならない耳だれが特徴です。中耳や内耳が破壊され難聴になることがあります。
-
造血器病変
赤血球や血小板が減り、貧血や出血をきたします。
-
肝・脾臓病変
肝臓や脾臓が腫れます。肝臓の働きが悪くなり、むくみや腹水、黄疸がでることがあります。
-
肺病変
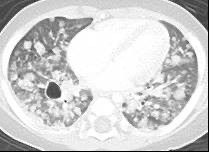
成人では肺だけに病変がある患者さんがあり、検診などでたまたま見つかることがあります。成人の患者さんのほとんどは喫煙者です。子どもでは、ほとんどが多臓器型で肺が病変のみという患者さんはありません。自覚症状としては、乾いた咳、息切れ、息苦しさです。肺が破壊されると、肺が空気のふくろにおきかわり、それが破れると、肺と胸の壁の間に空気が漏れて肺が縮んでしまう、すなわち気胸をおこします。さらに肺の破壊が進むと、空気のふくろだらけ、すなわち蜂巣様肺となり、息ができなくなることがあります。
-
消化管病変
口内炎や歯肉の腫れがおこることがあります。腸にも病変ができることがあり、下痢をしたり、便に血が混じったりすることがあります。
-
視床下部・下垂体病変
脳の奥のほうの視床下部と下垂体と呼ばれるところにしばしば病変が見られます。視床下部からは、下垂体を刺激するホルモンが出て、それによって下垂体からさまざまなホルモンが出ます。下垂体の後葉というところから、尿を濃くするホルモン、すなわち抗利尿ホルモン(ADH)が分泌されます。このADHの分泌が悪くなるのが中枢性尿崩症で、薄い尿が多量にでるためにのどが渇き多量に水分を飲む状態になります。LCHでは尿崩症が高率にみられます。LCHの診断時にすでに尿崩症が見られることもありますが、2-3年してからでてくることも多々あります。頭部MRIで下垂体後葉の高輝度スポットがなくなるのが特徴です。また、視床下部から下垂体への連絡路である下垂体茎が太くなることもよくあります。一度ADHの分泌が悪くなると通常は元に戻ることはありませんが、DDAVPという点鼻薬でホルモンを補充すれば尿量をコントロールすることができます。下垂体の前葉というところが障害されると、成長ホルモンや甲状腺刺激ホルモン、性腺刺激ホルモン、副腎皮質刺激ホルモンなどの分泌が悪くなり、背が伸びなくなったり、月経が起こらなくなったり、さまざまな症状が出ます。
視床下部が障害されると、ホルモンの分泌が悪くなるだけではなく、食欲の異常(極度の肥満や痩せ)、体温調節の異常、自律神経調節の異常(血圧や脈拍などの調節)などが出ることがあります。
-
脳病変
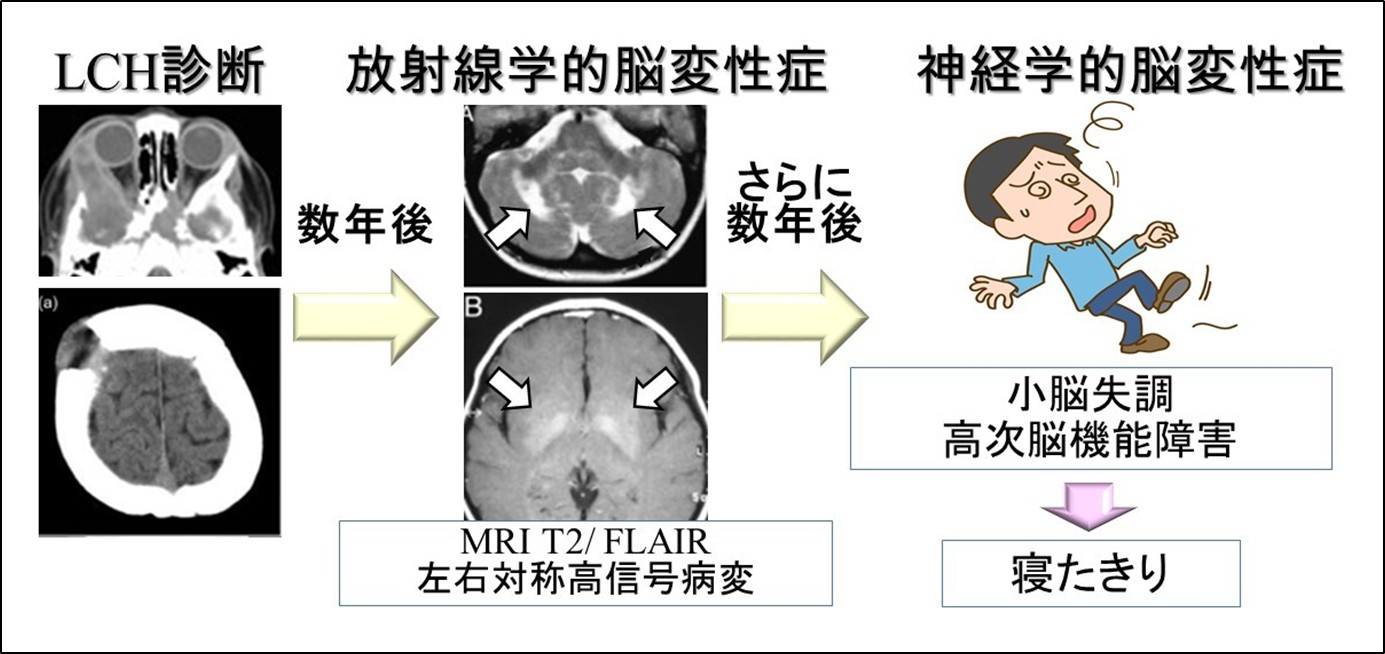
LCHの脳の病変には前述の視床下部・下垂体病変のほかに、脳そのものあるいは脳を包んでいる膜に腫瘤ができる病変、もうひとつは、脳変性病変です。
脳変性病変では、LCHの診断後数年して頭部MRIで小脳や大脳基底核という部分に左右対称に病変があらわれ(放射線学的脳変性症)、さらに数年して、転びやすい、うまくしゃべれない、飲み込みにくい、手が震える、集中できない、性格がかわるなどの症状がでてきて(神経学的脳変性症)、徐々に進行し最終的に寝たきりになります。前述のBRAF V600E変異のある患者さんがほとんどだと言われています。改善させる有効な治療法はわかっていませんが、BRAF阻害薬が有効であったという報告があります(保険未承認)。日本では進行を食い止めるために、γグロブリン療法が試みられています(保険未承認)。
後述のCNSリスク部位に病変がある患者さんでは、視床下部・下垂体病変や脳変性病変がでてくる頻度が高くなりますので、頭部MRIを定期的に受けて、病変の早期発見をすることが大切です。
どのように診断しますか?
LCHの症状はさまざまで、症状が出そろうまでに時間がかかることもあります。子どもにはありふれた症状ですが、皮疹や耳漏れがなかなか治らない場合、LCHを疑うことが大切です。診断の決め手になる血液検査はありませんが、炎症の指標である、赤血球沈降速度(赤沈)やCRP、可溶性IL-2受容体が高くなることが多いです。レントゲン検査やCT検査などによって、どこにどれだけ病変がひろがっているのかを知ることが重要です。
診断を確定するには、皮膚や骨などの病変の一部を採って顕微鏡で見て確かめること(生検による病理検査)が必要です。ピーナッツ様の核をした組織球が好酸球やリンパ球などとともに集まり、この組織球が免疫染色でCD1aまたはCD207(ランゲリン)陽性であれば診断は確定します。
治療はどうしますか?
どこにどれだけ病変があるかによって治療方法が違ってきます。単一臓器型で1か所にしか病変がない場合には何もしなくても自然によくなることもあり、治療をせず様子を見ることもあります。一方、多臓器型の場合は続発症がでたり、命にかかわったりすることもあり、抗がん剤を使った治療(化学療法)を行います。以下、病型別に治療法を示します。
| a) 骨の1か所の病変 | 手や足の骨の場合、自然によくなることも多く、生検をした時に病変部を削るまたは副腎皮質ホルモン(ステロイド剤)を注入する治療をします。 骨病変を大きく削り取る治療をすると、骨が再生せず骨が欠損したままとなるのでお薦めできません。化学療法の効果がでてくると骨は徐々に再生し融けていた骨が元の形に戻ってきます。 乳突蜂巣(耳の後ろ)や眼窩(目の周り)、頭蓋底(脳の底)、副鼻腔(鼻の奥)などの骨に病変がある場合、尿崩症の発症頻度が高くなると言われ、これらをCNSリスク部位と呼びます。CNSリスク部位の病変の場合、単一病変であっても化学療法が薦められます。 |
|---|---|
| b) 骨の複数の病変 | 続発症を少なくするために、化学療法を行います。 |
| c) 皮膚病変 | 自然によくなることも多い一方、多臓器型に進行することもあります。ステロイド剤の塗り薬を使い注意深く経過を見るか、または化学療法を行います。 |
| d) 肺病変 | 成人ではまず禁煙します。これだけでよくなることがあります。徐々に息苦しさが進むこともあり、そのような場合、ステロイド剤による治療が行われることがあります。子どもの場合、多臓器型の一部として肺病変が見られるので、化学療法が必要です。 |
化学療法が必須です。ステロイド剤とビンカアルカロイド(ビンブラスチンまたはビンクリスチン)の基本薬剤に、シンタラビン、6-メルカプトプリンなどを組み合わせた化学療法を約1年間行います(ビンブラスチンのみ保険承認)。欧米ではLCH-IV、日本ではLCH-19-MSMFBプロトコールによる多施設共同臨床研究が行われています。
化学療法の効果がなく、急速に進行するまたは病変が消えない患者さんには、クラドリビン(2-CdA)/シタラビン(Ara-C)療法(保険未承認)、同種造血幹細胞移植が試みられています。BRAF V600E変異のある患者さんにはベムラフェニブやダブラフェニブというBRAF阻害剤が有効ですが保険未承認です。
再発した患者さんには、クラドリビンや破骨細胞を抑制するビスフォスフォネート(保険未承認)などによる治療が試みられています。
以前は、エトポシド(VP-16)が治療薬の主役でした。しかし、ほかの薬より優れていることが証明できなかったこと、二次性白血病をおこす危険性があることより、一般的には使われなくなりました。また、放射線治療も、通常は行われなくなりました。
成人の患者さんは、発症から治療開始までに時間がかかっていることが多く、後述する続発症がすでにでていることが多々あります。子どもに比べ、病気の勢いは低いことが多いですが、治療の副作用が出やすく、治療に反応しにくいことも多々あります。よって、成人患者さんと小児患者さんを全く同じように治療することは困難です。小児の多臓器型に対する治療の維持療法(治療後半部分の弱めの治療)で治療する試みがされています。
経過はどうなっていきますか?
ほとんどのLCH患者さんは命にかかわることはありませんが、なかには全く治療の効果が見られず坂を転げ落ちるようにどんどん悪化することもあり、経過はさまざまです。 LCHの病変が、肝臓・脾臓または造血器にある患者さんは死亡率が高いため、この3つをリスク臓器と呼びます。①肝は3cm以上腫れているまたは肝機能不全がある、生検で確かめられたとき、②脾臓は2cm以上腫れているとき、③造血器は、貧血(ヘモグロビン10g/dl未満)または白血球数減少(4,000未満)、血小板減少(10万未満)があるとき、病変ありと診断します。以前リスク臓器に含めていた肺は、現在はリスク臓器とはしていません。
単一臓器型やリスク臓器に病変がない患者さんの死亡率はほぼ0%です。しかし、リスク臓器に病変がある多臓器型の患者さんで、治療開始後6週間で化学療法の治療効果がでない場合には、死亡率は30%に上ります。日本の小児LCHの臨床研究(JLSG-96/-02)では、多発骨型(複数の骨病変)とリスク臓器病変のない多臓器型の死亡率は0%、リス期臓器病変のある多臓器型では8%でした。欧米の成績と比べると、リスク臓器病変のある多臓器型の死亡率は極めて低くなっていました。
化学療法によって一旦症状が軽減またはほぼ消失しても再発する患者さんは、多発骨型で3分の1、多臓器型で約半数に上ります。治療が奏効し症状がなくなって3年以上経過していれば、その後に再発することはまれです。再発の部位としては骨が最も多いです。再発しても、治療によりよくなることがほとんどで、命にかかわることはきわめて稀です。しかし、再発した患者さんでは、尿崩症や脳変性症などの続発症がでる率が高くなります。
続発症にはどんなものが、どのくらいありますか?
LCHは経過が長い病気です。欧米からの報告では、10年間の経過の中で、多臓器型の患者さんの70%以上に何らかの続発症がでるといわれます。最も頻度が高いのは尿崩症で40%、次いで難聴、手足の骨や背骨の変形による整形外科的問題、神経障害、成長障害が20%前後にみられます。整形外科的問題は、多臓器型だけでなく骨単独でも複数の骨に病変のある患者さんは高率にみられます。そのほか、肺病変による慢性呼吸不全、2次性白血病の発生などがあります。
日本の小児LCHの臨床研究(JLSG-96/-02)では、続発症のでる率は多臓器型で約20%です。欧米からの報告よりも極めて低くなっていました。適切な化学療法によって続発症が減ると考えられます。
今後の課題は何ですか?
小児LCHについては、かなり治療法がわかってきました。今後、再発・続発症を減らすにはどうしたらよいか、治療に反応せず急速に進行する患者さんをどう救うかが課題です。成人LCHについては、どのような患者さんがどのくらいいてどのように経過していくのかが、まだよくわかっていないのが現状です。まずそれを明らかにし、どのような患者さんにどのような治療がいいのかを探っていく必要があります。
LCH細胞に発がん性の遺伝子変異が明らかになってきたので、変異した遺伝子から作られるタンパク質の働きを抑える薬(BRAF阻害薬など)を使うことによって、LCHを治療できる可能性があります。この種の薬は、悪性黒色腫に対してすでに使われていますが、LCHに対してはまだ十分なデータがなく、保険で認められていません。今後の薬の開発が期待されます。
もっと詳しく知りたい方への参考文献
- 森本 哲, 塩田曜子, 坂本謙一, 工藤 耕, 今村俊彦, 工藤寿子. ランゲルハンス組織球症における病態解明と治療の展望. 臨床血液 2022; 63: 373-382.
- 森本 哲. 組織球症 ランゲルハンス細胞組織球症. 日本小児血液・がん学会(編), 小児血液・腫瘍学 改訂第2 版, 東京, 診断と治療社, 2022: pp523-526.
- 坂本 謙一, 塩田 曜子, 森本 哲, 今宿 晋作. Langerhans 細胞組織球症関連中枢神経変性症. 日本小児科学会雑誌 2021; 125: 1524-1535.
- 森本 哲. ランゲルハンス細胞組織球症~その細胞起源と細胞活性化機構. 炎症と免疫 2020; 28: 106-112.
- 森本 哲, 塩田曜子, 工藤寿子, 今村俊彦.ランゲルハンス細胞組織球症.に対する化学療法の適応と有効性. 血液内科 2015; 71: 535-541.
- Morimoto A, Shioda Y, Sakamoto K, Imamura T, Imashuku S; Japan LCH Study Group. Bone lesions of Langerhans cell histiocytosis triggered by trauma in children. Pediatr Int. 2022;64(1):e15199.
- Sakamoto K, Morimoto A, Shioda Y, Imamura T, Imashuku S; Japan LCH Study Group (JLSG). Long-term complications in uniformly treated paediatric Langerhans histiocytosis patients disclosed by 12 years of follow-up of the JLSG-96/02 studies. Br J Haematol. 2021;192(3):615-620.
- Sakamoto K, Morimoto A, Shioda Y, Imamura T, Imashuku S; Japan LCH Study Group (JLSG). Central diabetes insipidus in pediatric patients with Langerhans cell histiocytosis: Results from the JLSG-96/02 studies. Pediatr Blood Cancer. 2019;66(1):e27454.
- Morimoto A, Shioda Y, Imamura T, Kudo K, Kawaguchi H, Sakashita K, Yasui M, Koga Y, Kobayashi R, Ishii E, Fujimoto J, Horibe K, Bessho F, Tsunematsu Y, Imashuku S. Intensified and prolonged therapy comprising cytarabine, vincristine and prednisolone improves outcome in patients with multisystem Langerhans cell histiocytosis: results of the Japan Langerhans Cell Histiocytosis Study Group-02 Protocol Study. Int J Hematol. 2016 Jul;104(1):99-109.
- Morimoto A, Shimazaki C, Takahashi S, Yoshikawa K, Nishimura R, Wakita H, Kobayashi Y, Kanegane H, Tojo A, Imamura T, Imashuku S. Therapeutic outcome of multifocal Langerhans cell histiocytosis in adults treated with the Special C regimen formulated by the Japan LCH Study Group. Int J Hematol. 2013 Jan;97(1):103-8.
- Imashuku S, Kudo N, Kaneda S, Kuroda H, Shiwa T, Hiraiwa T, Inagaki A, Morimoto A. Treatment of patients with hypothalamic-pituitary lesions as adult-onset Langerhans cell histiocytosis. Int J Hematol. 2011; 94(6): 556-560.
- Imashuku S, Shioda Y, Kobayashi R, Hosoi G, Fujino H, Seto S, Wakita H, Oka A, Okazaki N, Fujita N, Minato T, Koike K, Tsunematsu Y, Morimoto A. Neurodegenerative central nervous system disease as late sequelae of Langerhans cell histiocytosis. Report from the Japan LCH Study Group. Haematologica. 2008 Apr;93(4):615-8.
AMED ⾰新的がん医療実⽤化研究事業「組織球症の標準治療確⽴を⽬的としたレジストリおよびバイオレポジトリの構築」佐藤班
改訂履歴
- Ver.1: 2010/01
- Ver.2: 2022/10