








黄色肉芽腫Ver.1 2026/1
Xanthogranuloma:XG
はじめに
黄色肉芽腫(xanthogranuloma, XG)は、主に乳幼児にみられる、非ランゲルハンス細胞性組織球性腫瘍の中では最も多い組織球性腫瘍である。ほとんどの患者は小児なので、接頭語として若年性(juvenile)をつけて若年性黄色肉芽腫(JXG)と呼ばれることも多い。一方、成人の皮膚に限局する病型が約10%あり、これを成人XG(Adult XG: AXG)と呼ぶことがある。XGは主として皮膚に限局する良性病変として経過するが、一部は眼や中枢神経、肝臓、脾臓などに浸潤し重篤な臨床像を呈する。2016年にHistiocyte Societyから組織球症の新しい分類が提唱され1)、(1) Langerhans-related (L-Group), (2) cutaneous and mucocutaneous (C-Group), (3) malignanthistiocytoses (M-Group), (4)Rosai-Dorfman disease (R-Group), (5) hemophagocytic lymphohistiocytosis and macrophage activation syndrome (H-Group)に分けられているが、XGの皮膚限局型はC-Group、多臓器型はL-Groupに分類されている。2022年に改訂された第5版WHO分類では、JXGとして「組織球系腫瘍および樹状細胞性腫瘍」に分類されている3)。
病態
単球・マクロファージ系胞由来の異常細胞が皮膚や臓器に浸潤・蓄積することで発症する。従来は反応性疾患と捉えられてきたが、近年の分子生物学的研究により、異常細胞におけるMAPK 経路の遺伝子に活性化変異の存在が明らかとなり、炎症性骨髄系腫瘍と再定義されている2,4)。XGにおける遺伝子変異を図1に示す5)。
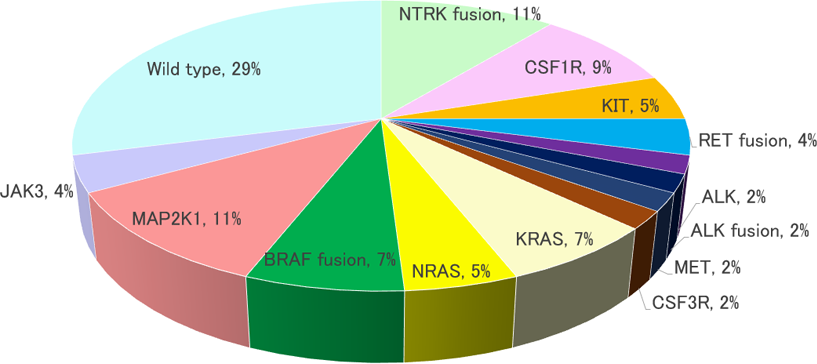
図1.黄食肉芽腫の遺伝子変異
(N=55, 文献5を改変)
L-Groupに属するLangerhans細胞組織球症(LCH)やErdheim-Chester病(ECD)と同様に、XGでも病的細胞にMAPキナーゼ経路の遺伝子に変異が認められるが、これらの約半数に認められるBRAF V600E変異はXGでは稀で(10%未満)ある。XGでは、LCHにおいては稀な、より上流に位置するチロシンキナーゼ受容体(CSF1R、NTRK、KITなど)や、その直下の細胞質内シグナル伝達分子(RASなど)に関連する遺伝子異常が多く報告されている。中でも、最近、NTRK融合遺伝子が高頻度に(30-50%)に認められることが明らかとなり、注目されている6)。また、チロシンキナーゼ受容体であるALK遺伝子が他の遺伝子と融合し、組織球性腫瘍を引き起こすことがあり、これはALK陽性組織球症として認識されXGとは別に分類する考えがあるが7)、臨床症状や病理組織学的所見はXGと鑑別困難である。
神経線維腫症1型(Neurofibromatosis type 1, NF1)の患者は、生来NF1遺伝子の機能喪失型変異を有するため、RASが持続的に活性化しており、その結果MAPキナーゼ経路が恒常的に刺激され、XGを発症しやすいと考えられている。また、若年性骨髄単球性白血病(JMML)は、NF1やRAS遺伝子などの異常によって発症することが知られているが、XGを合併する例もある8)。
病型・臨床症状
XGは以下の病型に分類される。病型別の頻度(図2)9)と皮膚限局型以外での病変臓器の頻度(図3)10)を図に示す。
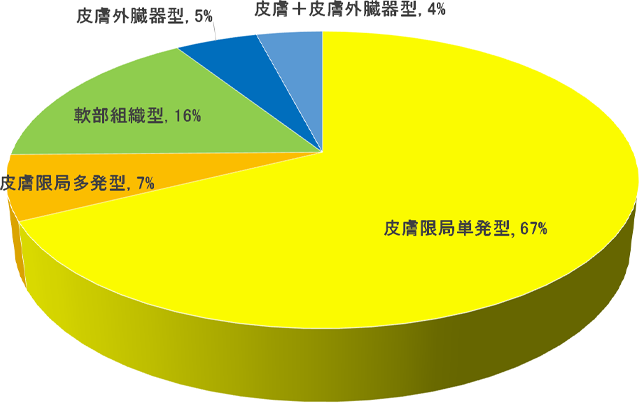
図2.病型別の頻度
(N=174,文献9を改変)
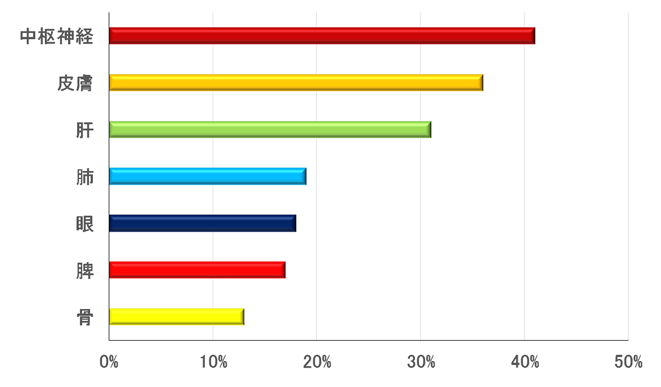
図3.皮膚限局型以外での病変臓器の頻度
(N=159, 文献10を改変)
-
皮膚限局型(cutaneous XG)
最も頻度が高く、半数は1歳までに出現する。頭部や体幹に、単発または多発の2-6mm大の橙赤色から黄褐色の丘疹・結節がみられ、Blueberry muffin rashとも呼ばれる。大半は無症候で、乳幼児では数年以内に自然消退する。成人では、ほとんどが単発性であるが、自然消退することは稀である。
-
眼病変(ocular XG)
乳児期に虹彩腫瘤として発症することが多く、結膜充血や前房出血、続発緑内障を呈する。放置すると失明に至る可能性がある。視機能予後に直結するため早期発見が重要である11)。
-
中枢神経型(CNS XG)
全身性病変の一部として認められる場合と、中枢神経(脳)に限局して発生する場合があり、全体の約1%と稀である。病変の局在に応じて、けいれん発作、視力障害、運動麻痺、頭蓋内圧亢進に伴う頭痛・嘔気・意識障害、学習障害に加え、尿崩症や成長ホルモン欠乏症などの内分泌異常を呈することがある。さらに、しばしば永続的な神経学的後遺症を残すことが知られている10)。
-
全身型(systemic XG)
全XG症例の1–5%に認められる稀な病態であり、肝臓、脾臓、肺、中枢神経系、骨髄、眼など多臓器に病変を生じる10,12)。臨床症状は病変部位に依存し、巨大な肝脾腫、血球減少、呼吸障害、神経症状、内分泌異常、視力障害など多彩である。生後6か月以内に発症することが多く、皮膚型よりも低年齢で発症する。原因不明の血球減少、肝臓障害、呼吸障害をきたした乳児では、全身性XGも疑う必要がある。致死率が高く、早期診断、治療介入が必要である10,13)。
多臓器型XG、特にBRAF V600E変異陽性の例は中枢神経の腫瘤病変を伴うことが多く、若年発症のECDとする考えがある13)。また、AXGは、他の血液悪性腫瘍を合併することがあり、同一cloneを起源とする可能性が示唆されている15)。
診断と検査
診断は病変組織を生検し病理学的に行う。泡沫状の組織球(xanthoma cell)や Touton 型多核巨細胞を特徴とするが、全例に見られるわけではない。免疫表現型はCD68陽性、CD163陽性、CD1a陰性、Langerin(CD207)陰性であり、LCHとの鑑別に重要である2,8)。病理像は、Early、Classic、Late(transitional)に分類される。Early typeは初期段階の病変で、小型の均一な組織球が密に浸潤しているが多核巨細胞は見られない。Classic typeは最も一般的なタイプで、泡沫状の組織球や特徴的なTouton型多核巨細胞がみられる。Late typeは深部に発生することが多く、渦巻き状の(線維性)組織球の増殖がみられる12)。
補助検査として以下が推奨される。
| 画像検査 | 超音波、MRI、CT、PET/CTによる全身評価。 |
|---|---|
| 眼科的検査 | スリットランプで虹彩腫瘤や前房出血を確認。 |
| 血液検査 | 血算・肝腎機能・炎症マーカー。 |
| 骨髄検査 | 血算に異常があるときは骨髄検査が必要。 |
| 分子検査 | 標的治療の選択につながる。 |
治療
-
皮膚限局型
多くは自然消退し、治療を必要としない。整容目的や診断的に外科切除を行うこともある。
-
眼型
ステロイド点眼または全身投与が用いられる。孤発性眼病変は外科的切除で良好な経過をとることがある。
-
中枢神経型
孤発性で完全切除可能な場合は外科的に摘出する。多発性の場合や、外科的切除が不可能な場合には、LCHの治療に準じてステロイド剤とビンカアルカロイド(ビンクリスチンやビンブラスチン)などを用いた抗がん剤治療を行う。分子標的薬(BRAF/MEK阻害薬など)の使用も検討される14)。
-
全身型
ステロイド単独では不十分なことが多く、ビンクリスチン、シタラビン、クラドリビンなど、LCH 治療に準じた化学療法が行われる。難治例に対しては分子標的薬(BRAF/MEK阻害薬など)の使用も検討される2,8,10)。BRAF V600E変異陽性の場合、日本では2023年に保険承認された「標準的な治療が困難なBRAF遺伝子変異を有する進行・再発の組織球症にBRAF阻害薬であるダブラフェニブ(dabrafenib)とMEK阻害薬であるトラメチニブ(trametinib)の併用療法」が使用可能である。BRAF V600E変異以外のMAPキナーゼ経路の遺伝子変異の場合にはMEK阻害剤、ALK遺伝子変異の場合にはALK阻害剤が有効である可能性がある(保険適応外)。
参考文献
- Emile J-F, Abla O, Fraitag S, et al. Revised classification of histiocytoses and neoplasms of the macrophage-dendritic cell lineages. Blood. 2016; 127: 2672–2681.
- Koh KN, Yoon SH, Kang SH, et al. Advancements in the understanding and management of histiocytic neoplasms. Blood Res. 2024; 59: 22.
- Ferry JA, Hill B, Hsi ED. Mature B, T and NK-cell, plasma cell and histiocytic/dendritic cell neoplasms: classification according to the World Health Organization and International Consensus Classification. J Hematol Oncol. 2024; 17: 51.
- Durham BH. Molecular characterization of the histiocytoses: Neoplasia of dendritic cells and macrophages. Semin Cell Dev Biol. 2019; 86: 62-76.
- Durham BH, Rodrigo EL, Picarsic J, et al. Activating mutations in CSF1R and additional receptor tyrosine kinases in histiocytic neoplasms. Nat Med. 2019; 25: 1839-1842.
- Fragneau R, Fraitag S, Kemps PG, et al. NTRK1-rearranged histiocytosis: clinicopathologic and molecular features. Blood Adv. 2025; 9: 3617-3628.
- Kemps PG, Picarsic JL, Emile JF. ALK-Positive Histiocytosis-A Distinct Histiocytic Entity Deserving Recognition. JAMA Dermatol. 2024; 160: 685-686.
- McClain KL, Bigenwald C, Collin M, et al. Histiocytic disorders. Nat Rev Dis Primers, 2021; 7: 73.
- Dehner LP. Juvenile xanthogranulomas in the first two decades of life: a clinicopathologic study of 174 cases with cutaneous and extracutaneous manifestations. Am J Surg Pathol. 2003; 27: 579-593.
- Zou T, Wei A, Ma H, et al. Systemic juvenile xanthogranuloma: A systematic review. Pediatr Blood Cancer. 2023; 70: e30232.
- Samara WA, Khoo CTL, Say EAT, et al. Juvenile xanthogranuloma involving the eye and ocular adnexa: Tumor control, visual outcomes, and globe salvage in 30 patients. Ophthalmology. 2015; 122: 2130-2138.
- Janssen D, Harms D. Juvenile Xanthogranuloma in Childhood and Adolescence. Am J Surg Pathol. 2005; 29: 21-28.
- Maeda M, Morimoto A, Shioda Y, et al. Long-term outcomes of children with extracutaneous juvenile xanthogranulomas in Japan. Pediatr Blood Cancer. 2020; 67: e28381.
- Picarsic J, Pysher T, Zhou H, et al. BRAF V600E mutation in Juvenile Xanthogranuloma family neoplasms of the central nervous system (CNS-JXG): a revised diagnostic algorithm to include pediatric Erdheim-Chester disease. Acta Neuropathol Commun. 2019; 7: 168.
- Kwan M, Yang CS, Nguyen CV. Solitary and multiple xanthogranulomas in adult patients: a single-center retrospective cohort study. Arch Dermatol Res. 2025; 317: 660.