








黄色肉芽腫ってどんな病気?Ver.1 2026/2
黄色肉芽腫(Xanthogranuloma:XG)
Frequently Asked Questions(FAQ)
はじめに
⻩⾊⾁芽腫(xanthogranuloma:XG)は、まれな「炎症性⾻髄腫瘍」です(「組織球症ってなに?」を参照)。WHO分類では「組織球性腫瘍および樹状細胞性腫瘍」に分類されています。乳児期に発症することが多く、半数の患者さんは1歳未満なので、若年性⻩⾊⾁芽腫(Juvenilexanthogranuloma:JXG)と呼ばれることがしばしばあります。しかし、成⼈にも発⽣することがあるので、ここでは「若年性」を付けずに「⻩⾊⾁芽腫」として解説します。また、別項⽬で述べる中⾼年に多いエルドハイム・チェスター病(Erdheim-Chester disease:ECD)とXGは、病変を顕微鏡で視ても区別がつかないので、「XGファミリー」としてまとめる考えもありますが、ECDの症状や病変部位はかなり特徴的でXGとは異なるので、ここでは別に扱います。⼀⽅、ALKという遺伝⼦に変異がある「ALK陽性組織球症」は、顕微鏡で視てXGと区別がつかないにもかかわらず、XGと独⽴して分類されることがありますが、症状や病変部位はXGとかなり似ていますので、ここではXGに含めます。
XGの患者さんの数はよくわかっていませんが、⽇本で1年間に新たに診断される患者さんの数は年間に20例ほどと考えられます。
若年性黄色肉芽腫(Juvenile Xanthogranuloma:JXG)については、組織球症ねっとのJXGのページ(現在作成中)も参照してください。
原因はなんですか?
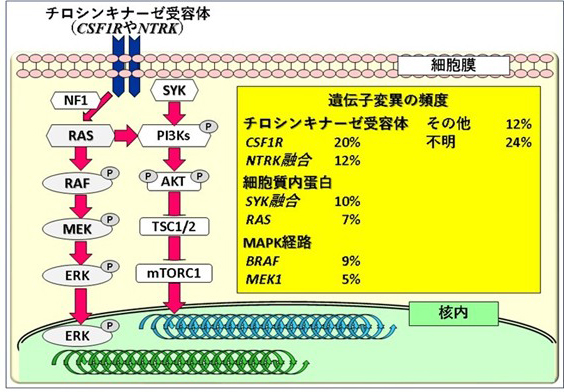
図1.XG細胞における遺伝子変異の頻度
LCHと同様に、⾻髄の造⾎前駆細胞(⽩⾎球や⾚⾎球、⾎⼩板の元になる細胞)に遺伝⼦変異が⼊り異常な組織球ができてしまうことが原因です。変異が⼊る遺伝⼦は、LCHと同様にMAPキナーゼ経路(「組織球症ってなに」を参照)の遺伝⼦(MAP2K1[MEK1と同義]やBRAF)もありますが、もっと上流のチロシンキナーゼ受容体(CSF1RやNTRK)や受容体のすぐ下流の細胞質内蛋⽩(SYKやRAS)の遺伝⼦が多いです(図1)。
神経線維腫症1型の患者さんは、⽣まれながらRASを不活化型にするNF1遺伝⼦に変異があるため、RASが常に活性化(スイッチオン)しています。よって、下流の経路が活性化し、XGを発症しやすいと⾔われています。また、若年性⾻髄単球性⽩⾎病は、NF1やRAS遺伝⼦などに変異が⼊ることによって発症しますが、同時にXGを発症することもあります。また、チロシンキナーセ受容体であるALK遺伝⼦が他の蛋⽩の遺伝⼦と融合して組織球性腫瘍が⽣じることがあり、ALK陽性組織球症として認識されていますが、症状も病変の組織所⾒もXGとは区別がつきません。
病型とその特徴
-
⽪膚限局型(⽪膚)XG
 若年性黄色肉芽腫(JXG)の場合、
若年性黄色肉芽腫(JXG)の場合、
皮疹のみで自然軽快する例が多いXG患者さんの70‐80%はこのタイプです。⾚みがかった、あるいは、⻩⾊または茶⾊がかった、わずかに盛り上がった数ミリ⼤の結節が⽪膚に⽣じます。頭部や⾸、胸や腹、背中などの最も多く発⽣します。ほとんどの場合、1〜5年以内に平坦化して⾃然に消えます。⽪膚の萎縮や⾊素沈着が残ることがあります。
-
眼XG
⾮常にまれな病変ですが、治療が遅れると失明する可能性があります。ほとんどの患者さんは、⽚側の眼にだけ病変が⽣じます。虹彩や結膜に病変が⽣じることが多く、眼球結膜(⽩⽬)の充⾎や前房(⾓膜とレンズの間)への出⾎が最も多い所⾒です。
-
中枢神経XG
中枢神経(脳)にだけ病変がある場合と、全⾝性病変の⼀部として発⽣する場合とがあり、XGの患者さんの約1%と稀な病変です。病変の部位によって、けいれん発作、視⼒障害、運動⿇痺、頭蓋内圧の上昇に伴う頭痛や吐き気・意識障害、学習障害、尿崩症や成⻑ホルモン⽋乏症などの内分泌異常など、さまざまな症状が現れます。神経学的な後遺症を伴うことがしばしばあります。
-
全身性XG
全⾝性XGは、XG患者さんの約4%を占めます。⽣後1か⽉くらいで発症することが多いです。肝臓、肺、軟部組織、中枢神経、脾臓など、さまざまな臓器に病変がみられますが、 ⽪膚病変を伴うことは稀です。症状は病変部位によってさまざまです。原因不明の⾎球減少、肝臓障害、呼吸障害をきたした乳児では、全⾝性XGも疑う必要があります。死亡率は5〜10%に昇ります。特に1か⽉未満で発症した場合には、臓器不全が進⾏し死亡率が⾼くなります。
診断と検査
病変の⼀部を採って顕微鏡で視て診断します。CD1aおよびCD207というLCHに特徴的な細胞の標識がない組織球がたくさん集まっています。初期には⽐較的⼩型の組織球が多くみられますが、典型的にはTouton型といわれる核がリング状に並んだ多核の巨細胞がみられ、時間が経つと紡錘形の細胞が⽬⽴つようになります。この顕微鏡所⾒はECDと同じで区別がつきません。XGと診断がついたら⾎液検査や眼科検査が必要です。貧⾎や⾎⼩板減少などがある場合には⾻髄検査が必要です。また、脳MRI検査や腹部超⾳波/CT検査などで病変が隠れていないか検査します。成⼈の場合、ECDでは両側の⼤腿⾻と脛の⾻に⾻硬化性病変が⾒られることが多いので、区別するのに役⽴ちます。成⼈では、⾎液悪性腫瘍を同時に発症する可能性があるため、注意が必要です。
治療
⽪膚に限局した患者さんのほとんどは治療の必要がありません。 眼の病変は、多くの場合ステロイド点眼薬で治療しますが、病変の切除やステロイド剤の内服を⾏うこともあります。 中枢神経の病変は、1か所だけで⼿術で取り除ける場合には、⼿術します。病変が複数あったり、⼿術で取り除けない部位であったりした場合には、LCHの治療に準じてステロイド剤とビンカアルカロイド(ビンブラスチンやビンブラスチン)を⽤いた抗がん剤治療を⾏います。全⾝性に病変がある場合にもLCHに準じた抗がん剤治療を⾏います。 難治性の場合には、クラドリビンやクロファラビンを⽤いることがあります。このような治療が効かない患者さんには、変異している遺伝⼦の働きを抑える分⼦標的療法が期待されます。BRAF V600E変異がある患者さん(XG患者さんの10%未満と少数)は、⽇本では2023年に保険承認された「標準的な治療が困難なBRAF遺伝⼦変異を有する進⾏・再発の組織球症にBRAF阻害薬であるダブラフェニブ(dabrafenib)とMEK阻害薬であるトラメチニブ(trametinib)の併⽤療法」を使うことができます。BRAF V600E変異以外の遺伝⼦変異がMAPキナーゼ経路にある場合にはMEK阻害剤、ALK遺伝⼦に変異がある場合にはALK阻害剤が役⽴つ可能性があります(いずれも保険適応外)。
もっと知りたい方への参考文献
- 坂本謙⼀. 若年性⻩⾊⾁芽腫症の診断・治療. 8 章 組織球症. ⼩児⽩⾎病・リンパ腫−Strategy & Practice. 中⼭書店. 2021, pp260-264.
- 佐藤亜紀, 坂本 謙⼀, 森本 哲. 【⾎液症候群(第 3 版)-その他の⾎液疾患を含めて-】リンパ系の腫瘍 組織球性疾患 腫瘍性組織球症 その他の組織球症(若年性⻩⾊⾁芽腫症,エルドハイム・チェスター病,Rosai-Dorfman-Destombes 病などの non-LCH). 2024; ⽇本臨床 別冊⾎液症候群 IV: 480-485.
- Dehner LP. Juvenile xanthogranulomas in the first two decades of life: a clinicopathologic study of 174 cases with cutaneous and extracutaneous manifestations. Am J Surg Pathol. 2003;27: 579593.
- Janssen D, Harms D. Juvenile xanthogranuloma in childhood and adolescence: a clinicopathologic study of 129 patients from the kiel pediatric tumor registry. Am J Surg Pathol. 2005; 29: 21-28.
- Stover DG, Alapati S, Regueira O, Turner C, Whitlock JA. Treatment of juvenile xanthogranuloma. Pediatr Blood Cancer. 2008; 51: 130-133.
- Maeda M, Morimoto A, Shioda Y, Asano T, Koga Y, Nakazawa Y, Kanegane H, Kudo K, Ohga S, Ishii E; Histiocytosis Study Group of the Japanese Society of Pediatric Hematology/Oncology. Long-term outcomes of children with extracutaneous juvenile xanthogranulomas in Japan. Pediatr Blood Cancer. 2020; 67: e28381.
- McClain KL, Bigenwald C, Collin M, Haroche J, Marsh RA, Merad M, Picarsic J, Ribeiro KB, Allen CE. Histiocytic disorders. Nat Rev Dis Primers. 2021; 7, 73.
- Kemps PG, Picarsic J, Durham BH, Hélias-Rodzewicz Z, Hiemcke-Jiwa L, van den Bos C, et al. ALK-positive histiocytosis: a new clinicopathologic spectrum highlighting neurologic involvement and responses to ALK inhibition. Blood. 2022; 139: 256-280.
- Kemps PG, Baelde HJ, Vorderman RHP, Stelloo E, Swennenhuis JF, Szuhai K, et al. Recurrent CLTC::SYK fusions and CSF1R mutations in juvenile xanthogranuloma of soft tissue. Blood. 2024; 144: 2439-2455.
AMED ⾰新的がん医療実⽤化研究事業「組織球症の標準治療確⽴を⽬的としたレジストリおよびバイオレポジトリの構築」佐藤班